





引 言
環(huán)氧樹脂[1](ER)是綜合性能優(yōu)良、應用廣泛 的熱固性樹脂,但也存在一些缺點,如固化物中存 在大量反應生成的羥基等極性基團,吸水率高、耐 濕性差、電性能不佳和固化樹脂脆性大等。為使 ER樹脂獲得高性能以滿足更高的應用要求,人們 不斷對ER進行改性,其中一條重要途徑就是通過 雙馬來酰亞胺(BMI)改性ER。
雙馬來酰亞胺(BMI)因其具有酰亞胺環(huán)結構而 具有優(yōu)異的熱穩(wěn)定性和力學性能[2],且與環(huán)氧樹 脂有良好的相容性[3]。因而近年來采用BMI改性 環(huán)氧樹脂研究引起了國內外學者的廣泛關注。劉柏 松[4]等人發(fā)現(xiàn),用BMI單體改性環(huán)氧樹脂、芳香 胺固化體系可以顯著提高環(huán)氧樹脂的韌性和耐熱 性。Dae SuKim[5]等人研究了BMI改性環(huán)氧樹脂體 系的固化行為,并對體系可能發(fā)生的化學反應進行 了探討。
二氮雜萘酮類雙酚單體(DHPZ)(見式1)是本 課題組近年來開發(fā)的1種新型類雙酚單體,具有不 對稱扭曲非共平面的結構,以及良好的耐熱性和可 溶性。PPEK是由DHPZ經與其他單體聚合得到的 一種新型高分子材料,本文通過在BMI中引入
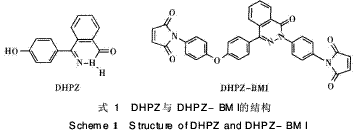
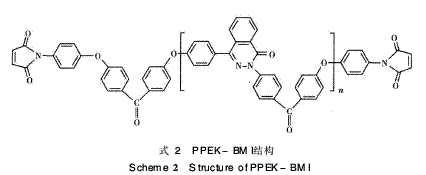
PPEK鏈段進行擴鏈,可進一步增大兩馬來酰亞胺 (M I)間R鏈的間距,從而適當增大鏈的自旋和柔軟 性,以達到降低固化物的交聯(lián)密度、減少鏈的剛性 及改善韌性的目的。因此本文采用自制的含二氮雜 萘酮聯(lián)苯結構雙馬來酰亞胺(DHPZ-BMI),即具有 耐高溫易溶解的雙馬來酰亞胺樹脂,與DDS形成組 合固化劑對E-51環(huán)氧樹脂進行固化改性,并將此 三元固化體系與鏈延長型含醚鍵雙馬來酰亞胺樹脂 PPEK-BMI(DP=15) /DDS/E-51固化體系作對 比,探討該體系的固化機理和熱穩(wěn)定性能(見式2)。
1 實驗部分
1·1 原 料
E-51環(huán)氧樹脂:雙酚A型,環(huán)氧值0·49~0·51; DHPZ-BMI:實驗室自制[6]; PPEK-BMI(DP=15): 實驗室自制; 4, 4′-二胺基二苯砜(DDS):化學純, 上海三愛思試劑有限公司生產。
1·2 DHPZ-BMI/DDS/E-51的制備
DHPZ-BMI/DDS以3∶1物質的量比混合均勻 后,再與E-51以1∶1質量比充分混合,置于 130℃油浴中攪拌30 min,得到酒紅色的粘稠狀產 物,室溫下自然冷卻。
1·3 固 化
將上述DHPZ-BMI/DDS/E-51置于真空烘箱 除氣,按150℃/1 h, 180℃/2 h, 200℃/4 h, 230℃/2 h的固化工藝進行固化,自然冷卻至室溫。
1·4 分析與表征
使用Nicolet-20DXB型傅里葉變換紅外光譜儀, 對固化過程進行跟蹤測試;使用NETZSCH DSC204示 差掃描量熱儀對樹脂固化過程的熱效應進行測定, N2氣氛,升溫速度分別為5, 10, 15℃/min;使 用NETZSCH TGA209型熱重分析儀對固化樹脂的 熱穩(wěn)定性進行分析, N2氣氛,升溫速度分別為5, 10, 15℃/min。
2 結果與討論
2·1 固化過程中的熱效應
圖1和圖2分別為DHPZ-BMI/DDS/E PPEK-BMI/DDS/E-51體系固化進程的曲線 可以看出:固化反應的反應放熱峰的位 溫速率密切相關,隨著升溫速率的提高,體 起始溫度和峰值溫度均提高,固化時間縮短 位時間產生的熱效應增大,固化反應放熱峰 向高溫移動。
對圖1采用Kissinger[7]方程計算固化反 化能與頻率因子。假設固化反應最大速率發(fā) 化反應放熱峰的峰頂溫度,反應級數(shù)在固化
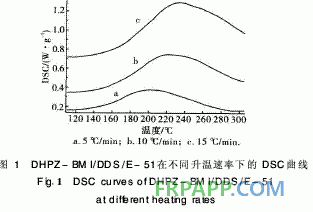

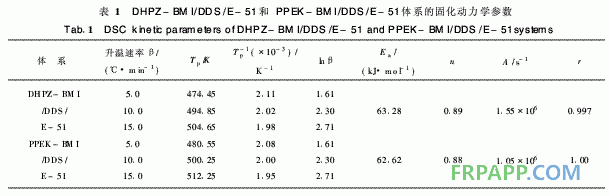
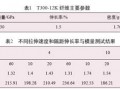
DHPZ-BMI/DDS/E-51體系的DSC數(shù)據(jù)及 固化反應動力學參數(shù)匯總見表1。同樣采用Kissi ger方程和Crane方程對圖2計算其固化反應表觀 活化能、表觀頻率因子、線性相關系數(shù)和反應級 數(shù)。由表1可見DHPZ-BMI/DDS/E-51和PPE -BMI/DDS/E-51兩種體系的固化反應活化能幾 乎相同,說明二者的固化反應過程相同,且固化反 應活化能均較低、頻率因子較大,故二者固化反應 速率均較快,與兩馬來酰亞胺(MI)間R鏈的間距 無關。
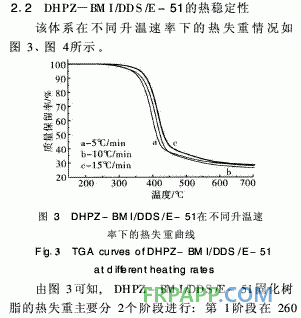
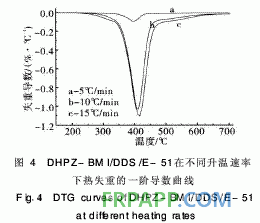
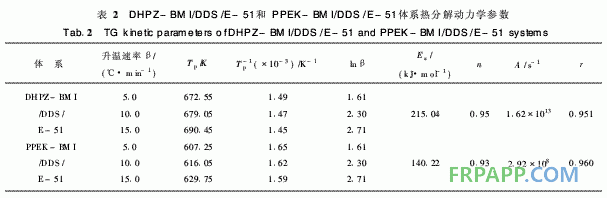
~440℃之間,起始分解溫度為260℃, 5%熱失 重溫度為341℃,該階段的熱分解失重可能是分子 間的弱鍵,如C—O—C的分解;第2階段在440℃ 之后,這一階段失重則是由于苯環(huán)、酰亞胺等裂解 造成的,且失重率明顯減小,再次證明固化體系良 好的熱穩(wěn)定性。從圖4取DTG曲線第1個峰的峰 頂溫度進行比較。按2·1節(jié)中的方法求取的熱分解 動力學參數(shù)見表2。
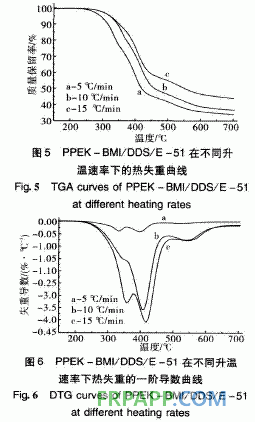
由圖5可知,其熱失重也分為2個階段:第 1階段在240~420℃之間,起始溫度為240℃, 5%熱失重溫度為298℃;第2階段在420℃之 后,失重率也明顯減小。從圖6取固化樹脂的 DTG第一峰頂溫度依Kissinger和Crane方程進行 計算,得到的固化樹脂的熱分解動力學分解參數(shù) 如表2所示。
表2中2種固化樹脂的熱分解活化能存在明顯 差異, DHPZ-BMI/DDS/E-51固化樹脂的分解活 化能較高,為PPEK-BMI/DDS/E-51固化樹脂的 1·5倍以上,這是因為DHPZ-BMI/DDS/E-51固 化樹脂中二氮雜萘聯(lián)苯結構比例高,耐熱性好,而 PPEK-BMI/DDS/E-51固化樹脂中有大量醚鍵存 在,降低了二氮雜萘聯(lián)苯結構在固化樹脂中的比 例,導致耐熱性下降,因此其起始分解溫度低,相 同熱失重的分解溫度也較低。
3 結 論
1)由DHPZ-BMI/DDS/E-51和PPEK-BMI/ DDS/E-51體系的固化反應動力學數(shù)據(jù)相比得知, 這2種體系的表觀活化能幾乎相同,說明二者的固 化反應過程相同,且固化反應活化能均較低、頻率 因子較大,故二固化反應速率均較快,與兩馬來酰 亞胺(MI)間R鏈的間距無關。
2)DHPZ-BMI/DDS/E-51和PPEK-BMI/ DDS/E-512種固化樹脂的熱分解活化能存在顯著 差別, DHPZ-BMI/DDS/E-51固化樹脂的熱分解 活化能達215·04 kJ/mo,l是PPEK-BMI/DDS/E- 51固化樹脂的1·5倍以上,這是因為DHPZ- BMI/DDS/E-51固化樹脂中二氮雜萘聯(lián)苯結構比 例高,耐熱性好,而PPEK-BMI/DDS/E-51固化 樹脂中有大量醚鍵存在,降低了二氮雜萘聯(lián)苯結構 在固化樹脂中的比例,導致耐熱性下降。亦即表 明:采用DHPZ-BMI/DDS組合固化劑固化的E- 51型環(huán)氧樹脂熱穩(wěn)定性更佳。







 魯ICP備2021047099號
魯ICP備2021047099號